Definition des Begriffs: Was ist FDA 21 CFR Part 11?
FDA 21 CFR Part 11 ist eine Reihe von Vorschriften, die 1997 von der US-amerikanischen Food and Drug Administration (FDA) eingeführt wurden. Diese Vorschriften definieren die Kriterien, unter denen elektronische Aufzeichnungen und elektronische Signaturen als vertrauenswürdig, zuverlässig und gleichwertig zu Papieraufzeichnungen und handschriftlichen Signaturen gelten. Im Wesentlichen legt Part 11 die Regeln für die Verwendung elektronischer Systeme in von der FDA regulierten Branchen fest, wie zum Beispiel Pharmazie und Biotechnologie.
Die Einführung von Part 11 zielte darauf ab, Prozesse zur Aufzeichnung und Verwaltung von Daten in den Bereichen Pharmazie und Life Sciences zu modernisieren und zu rationalisieren. Dabei sollte die Technologie genutzt werden, um gleichzeitig Datensicherheit und -integrität zu gewährleisten. Der Bedarf an solchen Vorschriften entstand, als diese Branchen vermehrt elektronische Methoden für die Dokumentation einführten.
Hauptanforderungen von FDA 21 CFR Part 11
- Elektronische Signaturen: Eine der zentralen Komponenten von FDA 21 CFR Part 11 ist die Verwendung elektronischer Signaturen. Die Anforderungen für elektronische Signaturen werden festgelegt, um deren Gleichwertigkeit mit handschriftlichen Signaturen zu gewährleisten. Dies umfasst die Gewährleistung der Sicherheit und Authentizität dieser Signaturen.
- Audit Trails: FDA 21 CFR Part 11 schreibt die Erstellung von Audit Trails für elektronische Aufzeichnungen vor. Diese Audit Trails dienen als chronologische Aufzeichnung von Systemaktivitäten, die Rückverfolgbarkeit und Verantwortlichkeit gewährleisten.
- Validierung elektronischer Systeme: Unternehmen müssen ihre elektronischen Systeme validieren, um sicherzustellen, dass sie den Anforderungen von Part 11 entsprechen. Dazu gehören umfangreiche Tests und Dokumentationen, um die Zuverlässigkeit und Genauigkeit der Systeme zu belegen.
- Zugangskontrollen: FDA 21 CFR Part 11 betont ebenfalls Zugangskontrollen, um sicherzustellen, dass nur autorisierte Personen Zugang zu elektronischen Aufzeichnungen und Systemen haben. Dies verbessert die Datensicherheit und verhindert unbefugte Änderungen.
Ihr Weg in die Digitalisierung - Entdecken Sie unsere Software
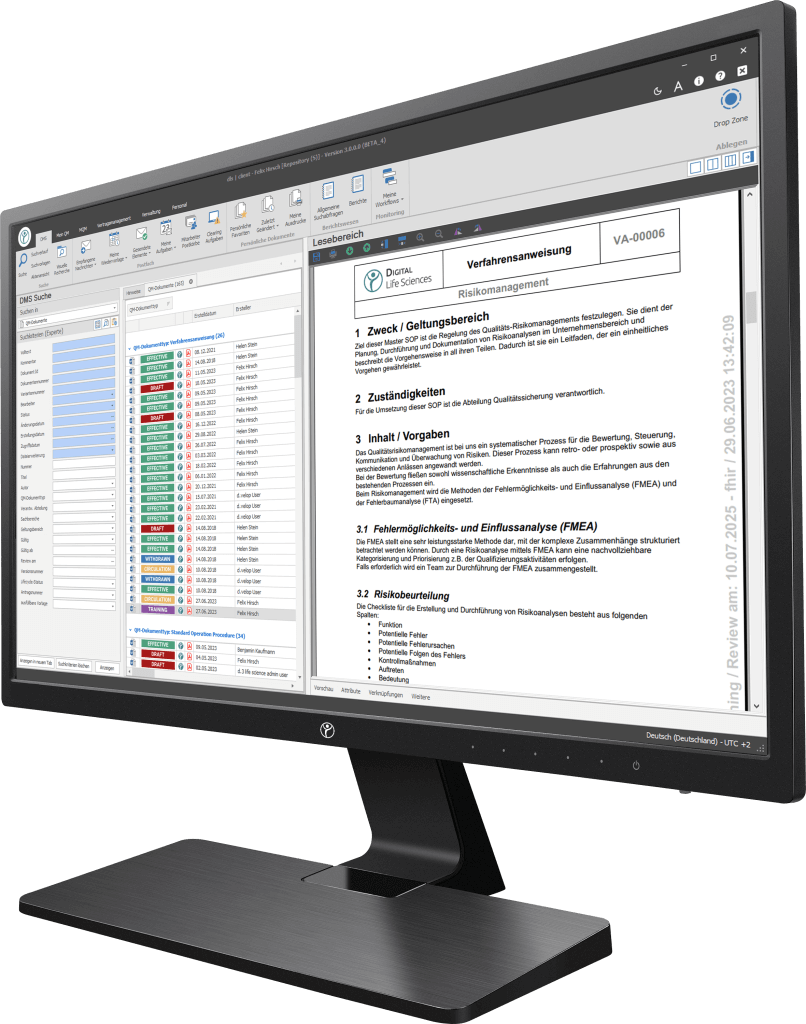
Unsere Digitalisierungslösungen adressieren in erster Linie dokumentenbasierte Prozesse der Herstellung, Produktion und des Qualitätsmanagements. Die Basis des dls | eQMS bildet ein ganzheitliches ECM/DMS-System. Das ECM/DMS-System kann an Ihr bestehendes ERP-System (z. B. SAP) angebunden werden und somit nahezu sämtliche dokumentenbasierte Prozesse im Unternehmen abbilden.
Einhaltung erreichen
Schritte zur Erreichung der Einhaltung von FDA 21 CFR Part 11:
- Bewertung: Beginnen Sie damit, Ihre aktuellen Systeme und Prozesse zu bewerten, um Compliance-Lücken zu identifizieren.
- Validierung: Stellen Sie sicher, dass Ihre elektronischen Systeme validiert sind und alle Software ordnungsgemäß getestet und dokumentiert sind.
- Zugangskontrolle: Implementieren Sie strenge Zugangskontrollen, um elektronische Aufzeichnungen zu schützen.
- Schulung: Schulen Sie Ihr Personal zu den Anforderungen von Part 11 und den besten Praktiken für die elektronische Dokumentation.
- Audit Trails: Legen Sie umfassende Audit Trails für elektronische Aufzeichnungen fest.
- Dokumentation: Führen Sie detaillierte Dokumentationen aller Prozesse und Systemvalidierungen durch.
Vorteile der Einhaltung
Die Einhaltung von FDA 21 CFR Part 11 bietet zahlreiche Vorteile:
- Verbesserte Datensicherheit
- Erhöhte Integrität und Authentizität der Daten
- Vereinfachte Aufzeichnungsführung
- Geringeres Risiko von behördlichen Verstößen
- Steigerung der Betriebseffizienz
Fazit
In einem digitalen Zeitalter, in dem Daten von zentraler Bedeutung sind, spielt die FDA 21 CFR Part 11 eine entscheidende Rolle bei der Gewährleistung der Integrität und Sicherheit elektronischer Aufzeichnungen in den Branchen Pharmazie und Life Sciences. Durch die Einhaltung der Anforderungen können Unternehmen nicht nur regulatorische Standards erfüllen, sondern auch ihre Datenverwaltungspraktiken stärken, was letztendlich zu sichereren und effizienteren Betriebsabläufen führt.
Starten Sie Ihre digitale Transformation mit unseren leistungsstarken, modularen Softwarelösungen
FAQ - FDA 21 CFR Part 11
Häufig gestellte Fragen (FAQs) zum FDA 21 CFR Part 11
FDA 21 CFR Part 11 gilt hauptsächlich für die Pharmazie-, Biotechnologie- und Medizinprodukteindustrie.
Ja, die Einhaltung von FDA 21 CFR Part 11 ist für Organisationen, die in dessen Anwendungsbereich tätig sind, obligatorisch.
Die Nicht-Einhaltung kann zu behördlichen Maßnahmen führen, einschließlich Geldbußen und Produkt-Rückrufen.
Ja, es gibt Softwarelösungen, die darauf abzielen, Unternehmen bei der Erreichung und Aufrechterhaltung der Einhaltung von Part 11 zu unterstützen. Einige dieser Tools finden Sie hier.
Audit Trails sollten regelmäßig überprüft werden, wobei die Häufigkeit von der Risikobewertung des Unternehmens und den Einhaltungsrichtlinien bestimmt wird.
Elektronische Systeme müssen sicherstellen, dass elektronische Aufzeichnungen und Unterschriften sicher, zuverlässig und vertrauenswürdig sind. Dies umfasst Zugangskontrollen, Prüfpfade, und die Validierung des Systems gemäß Vorschriften, die Integrität und Authentizität der Daten gewährleisten.
Ein elektronisches Qualitätsmanagementsystem (eQMS) erleichtert die Einhaltung von Compliance-Vorgaben durch automatisierte Überwachung und Dokumentation von prozessrelevanten Aktivitäten. Es bietet Funktionen wie CAPA-Management, Änderungs- und Abweichungskontrolle sowie die Verwaltung von SOPs und anderen relevanten Dokumenten.
Ein eDMS sollte eine Vielzahl von Aufzeichnungen verwalten, darunter Produktionsaufzeichnungen, Prüfprotokolle, abweichende Berichte, CAPA-Dokumentationen, Schulungsnachweise, Lieferantendokumentationen und Validierungsberichte. Diese Dokumentationen sind entscheidend für die Einhaltung gesetzlicher Bestimmungen und Qualitätsstandards.
Die Life-Science-Branche unterliegt strengen regulatorischen Anforderungen und Qualitätsstandards. Softwarelösungen zur Dokumentation und Compliance bieten effiziente und transparente Prozesse, die eine lückenlose Dokumentation, Kontrolle und Nachverfolgbarkeit sicherstellen. Dies ist unerlässlich, um den regulatorischen Anforderungen gerecht zu werden und die Produktqualität sowie -sicherheit zu gewährleisten.

