Definition of the term: What is FDA 21 CFR Part 11?
FDA 21 CFR Part 11 is a set of regulations introduced by the US Food and Drug Administration (FDA) in 1997. These regulations define the criteria under which electronic records and electronic signatures are considered trustworthy, reliable and equivalent to paper records and handwritten signatures. Essentially, Part 11 sets out the rules for the use of electronic systems in industries regulated by the FDA, such as pharmaceuticals and biotechnology.
The introduction of Part 11 was aimed at modernizing and streamlining processes for recording and managing data in the pharmaceutical and life sciences sectors. The technology should be used to ensure data security and integrity at the same time. The need for such regulations arose when these industries increasingly introduced electronic methods for documentation.
Main requirements of FDA 21 CFR Part 11
- Electronic signatures: One of the central components of FDA 21 CFR Part 11 is the use of electronic signatures. The requirements for electronic signatures are defined in order to ensure their equality with handwritten signatures. This includes ensuring the security and authenticity of these signatures.
- Audit trails: FDA 21 CFR Part 11 requires the creation of audit trails for electronic records. These audit trails serve as a chronological record of system activities that ensure traceability and accountability.
- Validation of electronic systems: Companies must validate their electronic systems to ensure that they meet the requirements of Part 11. This includes extensive tests and documentation to prove the reliability and accuracy of the systems.
- Access controls: FDA 21 CFR Part 11 also emphasizes access controls to ensure that only authorized individuals have access to electronic records and systems. This improves data security and prevents unauthorized changes.
Your path to digitization - Discover our software
Our digitization solutions primarily address document-based processes in manufacturing, production and quality management. The basis of the dls | eQMS is a holistic ECM/DMS system. The ECM/DMS system can be connected to your existing ERP system (e.g. SAP) and thus map almost all document-based processes in the company.
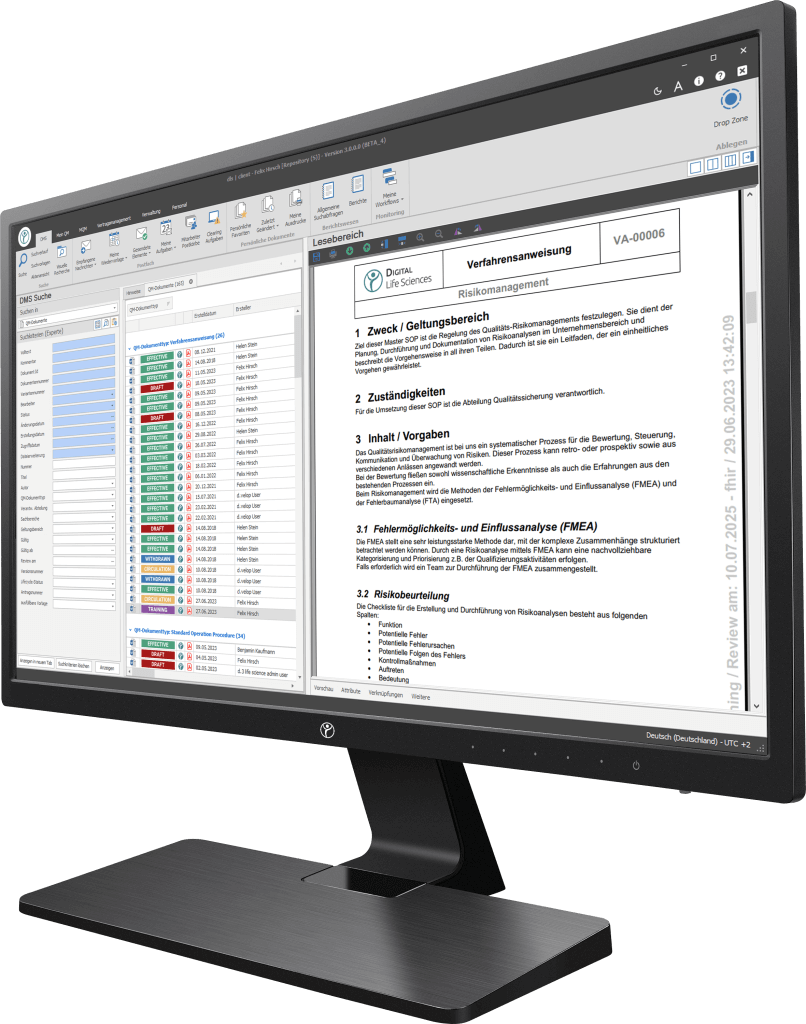
Achieve compliance
Steps to achieve compliance with FDA 21 CFR Part 11:
- Evaluation: Start by evaluating your current systems and processes to identify compliance gaps.
- Validation: Ensure that your electronic systems are validated and that all software is properly tested and documented.
- Access control: Implement strict access controls to protect electronic records.
- Training: Train your staff on Part 11 requirements and best practices for electronic documentation.
- Audit trails: Establish comprehensive audit trails for electronic records.
- Documentation: Carry out detailed documentation of all processes and system validations.
Advantages of compliance
Compliance with FDA 21 CFR Part 11 holds numerous advantages:
- Improved data security
- Increased data integrity and authenticity
- Simplified recording management
- Lower risk of regulatory violations
- Increase in operating efficiency
Conclusion
In a digital age where data is central, FDA 21 CFR Part 11 plays a critical role in ensuring the integrity and security of electronic records in the pharmaceutical and life sciences industries. By complying with the requirements, organizations can not only meet regulatory standards, but also strengthen their data management practices, ultimately leading to more secure and efficient operations.
Start your digital transformation with our powerful, modular software solutions
Frequently Asked Questions (FAQs)
To which industries does FDA 21 CFR Part 11 apply?
FDA 21 CFR Part 11 applies primarily to the pharmaceutical, biotechnology and medical device industries.
Is compliance with Part 11 mandatory?
Yes, compliance with FDA 21 CFR Part 11 is mandatory for organizations operating within its scope.
What are the consequences of non-compliance with FDA 21 CFR Part 11?
Non-compliance may result in regulatory measures, including fines and product recalls.
Are there special software solutions that have been developed for Part 11 compliance?
Yes, there are software solutions aimed at helping companies achieve and maintain Part 11 compliance. You can find some of these tools here.
How often should audit trails be checked?
Audit trails should be reviewed regularly, with the frequency determined by the organization’s risk evaluation and compliance policies.
What requirements must electronic systems fulfill in order to be compliant with FDA 21 CFR Part 11?
Electronic systems must ensure that electronic records and signatures are secure, reliable and trustworthy. This includes access controls, audit trails, and validation of the system according to regulations that ensure the integrity and authenticity of the data.
How does an eQMS support adherence to compliance requirements in pharmaceutical production?
An electronic quality management system (eQMS) facilitates adherence to compliance requirements through automated monitoring and documentation of process-relevant activities. It offers functions such as CAPA management, change and deviation control as well as the management of SOPs and other relevant documents.
What types of records should be managed in an electronic document management system (eDMS) for the food and pharmaceutical industry?
An eDMS should manage a variety of records, including production records, inspection records, deviation reports, CAPA documentation, training records, supplier documentation and validation reports. This documentation is crucial for compliance with legal regulations and quality standards.
Why is the provision of software solutions for documentation and compliance particularly important for the life sciences industry?
The life science industry is subject to strict regulatory requirements and quality standards. Software solutions for documentation and compliance offer efficient and transparent processes that ensure seamless documentation, control and traceability. This is essential in order to meet regulatory requirements and ensure product quality and safety.

