TECHNICAL DOCUMENTATION SOFTWARE
Digital software for your Technical Documentation
The Technical Documentation enables comprehensive digital management and updating of all documents. This ensures compliance with regulations such as the MDR and at the same time increases the efficiency and data security of your documentation-based processes.
Manage technical documents efficiently and securely
Compliance with the Medical Device Regulation (MDR)
Digitise processes and minimise lead times

You are currently viewing a placeholder content from YouTube. To access the actual content, click the button below. Please note that doing so will share data with third-party providers.
More Information» dls | technical documentation «
How does Technical Documentation support the documentation?
Software for your Technical Documentation
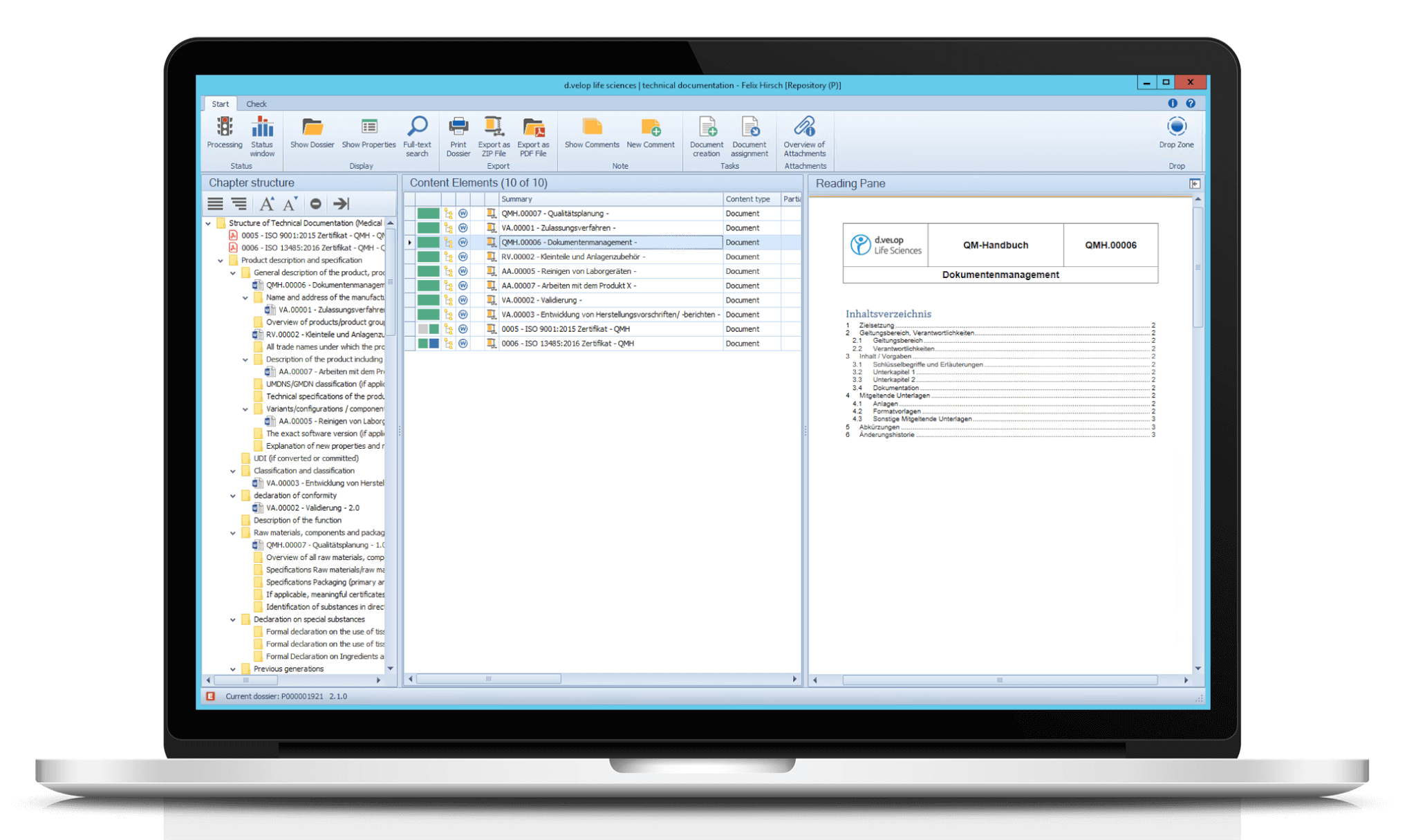
From individual documents to our document management system and process-oriented dossier management – Our module “dls | technical documentation” effectively supports the creation, updating and management of your technical documentation, e.g. in the context of the requirements of the Medical Device Regulation (MDR). Our process-oriented dossier management can be used in a variety of ways, e.g. to create site master files, tool logbooks, drug master files, QM documentation, project folders, validation documentation, and much more.
What are the benefits of Technical Documentation?
How do you benefit from the Technical Documentation software?
Digital management
Make manual documentation of dossiers a thing of the past – instead, manage them completely digitally.
Structured data maintenance
Let untidy file servers with multiple storage locations and redundant folder structures be a thing of the past.
Increased data security
Protect your data from unwanted interference by unauthorised users and from destructive forces.
Individual configuration
Let the modules be configured according to your specific requirements to facilitate the onboarding training of your colleagues.
Compliance with Medical Device Regulation
Create and update your technical documentation digitally to meet the requirements of the MDR.
Minimisation of runtimes
Reduce process runtimes for creating, revising and approving your dossiers and minimise the time spent searching for and compiling up-to-date documents.
Central information platform
Use d.velop documents (formerly d.3ecm) as your central source of information to retrieve all relevant data at any time.
Constant availability
Access your data anytime and anywhere.
Technical Documentation software demo
Digitalise your processes for greater efficiency
How do I work with the Technical Documentation software?
Discover some features of the Technical Documentation
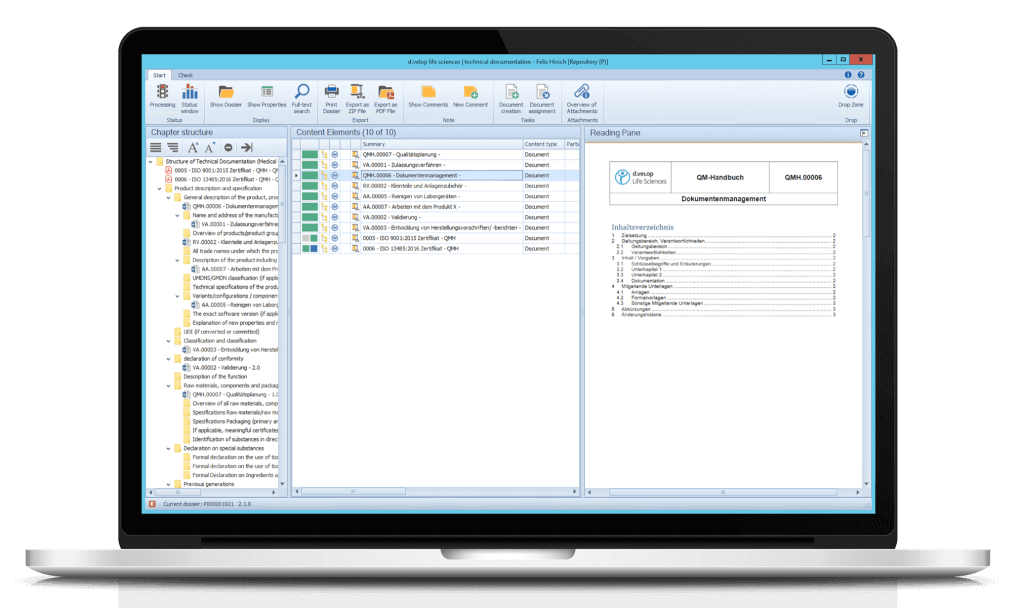
Moreover, you can directly adopt documents from the eDMS/eQMS using the dossier template. Add additional documents and attachments to the dossier application and assign them to the respective chapters via drag & drop.
Print your dossiers or export them as a ZIP file or PDF/A. It is also possible to print and/or export only parts or certain chapters of the dossier.
Using the Drop Zone, non-controlled and controlled documents can be stored via drag & drop. Documents can also be assigned directly to the respective nodes/chapters in the dossier via drag & drop.
For each document, a preview can be displayed in the flexibly customisable reading pane.
Create templates and chapter structures for your dossiers according to your needs and define responsibilities for each part. Dossier templates, e.g. for the MDR tree structure, are automatically included when the module is implemented.
What does Technical Documentation software look like live?
Would you like a live insight into the software?
Get a live insight into the possibilities of process-oriented dossier management in just 45 minutes using a sample use case. Learn how our solution can support and simplify the creation and updating of your technical documentation within the requirements of the Medical Device Regulation (MDR).
What functions does Technical Documentation software have?
Here are some features of the Technical Documentation
- Template management for various dossier structures
- Task management for processing individual chapters of a dossier (workflow)
- Seamless integration into our Document Control module
- Check the completeness of your dossiers
- Control of the current editing status of your dossier
- Versioning of your dossiers and easy access to all old versions
- Create comments with different priorities
- Export of the entire dossier or selected parts of it to a PDF or ZIP file
- Print of the entire dossier or selected parts of it
- Integrated electronic and GxP-compliant signature as well as an audit trail feature
- Classification of dossiers via attributes in ECM d.velop documents (formerly d.3ecm)
What regulations must Technical Documentation comply with?
Regulatory requirements for Technical Documentation
ISO 13485 MPG
Medical Device Regulation (MDR) - EU Directive MDR 2017/745
33 Good reasons to work with DLS
You are not convinced yet? Find out about 33 good reasons speaking for a cooperation with Digital Life Sciences GmbH. We will show you reasons from the provider’s point of view, from the software point of view and other general reasons that distinguish us.
Which companies use Technical Documentation?
Customer reviews of Technical Documentation
"We have been working together with d.velop AG very successfully and contentedly for years. Looking for a reliable partner for the Technical Documentation of our medical products, we stumbled upon the Life Sciences Client. With the transition to the new structure of EU MDR, we were able to fully exploit the potential of the Life Sciences Client for us and thus facilitate the work of document creation and improve acceptance in the company."

Which software can be used together with Technical Documentation software?
You might also like...
The Technical Documentation is a component of the Digital Life Sciences solution suite. Each product is powerful on its own, but when used together they are even better.

Document Control software
Whether work / process instructions (SOPs), process descriptions, test specifications or other document types — you can create, revise, and sign them all digitally with the document control software.

Training Management software
Extend the module “Document Control” to actively plan and log your employees' qualifications with our training management software.
QM processes (Complaint | DC | CAPA | CC)
Digitalise your ISO processes. Control your production-related QM processes using digital workflows.
Technical Documentation FAQ
Frequently asked questions (FAQs) about Technical Documentation
- Are there templates for the structure of the dossier?
Templates are available for frequently used documentation, e.g. according to Annex II of the MDR 2017/745. On the basis of a structure used by you, you can also amend templates yourself or have them created as part of the introduction of the module.
- Can chapter structures be customised?
You can customise chapter structures and dossier templates or create your own.
- Are there templates or examples for the Technical Documentation?
Many providers of documentation software offer comprehensive templates and examples to make the documentation process efficient. These include:
Technical Documentation templates: They are already structured and contain frequently used fields that ensure consistent and standard-compliant document creation.
Word templates for Technical Documentation: These templates are particularly suitable for smaller projects and allow easy customisation to specific requirements.
Examples of Technical Documentation in PDF format: These serve as a valuable guide in terms of structure and content and help users to create high-quality documents.
- How do I ensure that an employee adds all relevant documents to the dossier?
Using the tasks ‘Placeholders’ and ‘Document creation’ you can specify which documents the dossier must contain before it can be released.
- Can I use documents from the file system in the dossier?
Documents from your desktop or from the file directory can be used in the dossier. They are stored in the document management system and used from there in one or more dossiers.
- Can several employees work on one dossier?
For each dossier, a main person responsible is identified, who also receives the reminders for completion or periodic review. Individual chapters of the structure can be transferred to a person partially responsible for processing.
- Can documents be used more than once?
Documents are added once to the contents list of the dossier and can then be inserted at several positions in the structure. Documents can also be used in several dossiers.
- Why does one use the Technical Documentation for the approval of a medical device?
Technical Documentation is used for the approval of a medical device to provide regulatory authorities and others involved with information about how the device works, who will use it, and how it can be used safely.
- How can our software help you with Technical Documentation?
Our Technical Documentation software is an application primarily used for managing technical documentation (“dossiers”). It can manage a large number of dossiers in the process. The software allows you, for example, to create technical documentation in accordance with MDR (EU Medical Devices Regulation). The goal here is the audit-proof management of all documents and content that arise in the life of a medical device.
We will be happy to show you how you as a user can benefit from our software for technical documentation in your company in the future in a live webcast on the software.
- What are the key aspects of managing documentation software for medical devices?
The following aspects are of crucial importance when managing documentation for medical devices:
- The software should support comprehensive functions such as versioning, a release process and complete software documentation.
- Compliance with regulatory requirements, including technical documentation requirements and software documentation guidelines, is essential to ensure conformity and quality.
- What requirements must Technical Documentation for medical devices fulfill?
The requirements for technical documentation for medical devices include, among other things:
- Complete and transparent presentation of product information, e.g. in accordance with the Medical Device Regulation (MDR).
- Seamless integration of product documentation for medical devices and specific test reports.
- Ensuring traceability, e.g. through the use of templates for technical documentation.
- Use of a special software documentation template to meet regulatory requirements.
- How can you create technical documentation and what tools are available for this?
Various approaches are available to you for creating technical documentation:
- Manual creation using specific Word templates for technical documentation.
- Use of specialised software solutions for technical documentation that offer ready-made templates and comprehensive structuring options to meet the documented processes.
- Creation of technical documentation in PDF formats to ensure easy distribution and long-term archiving.
Furthermore, training plays an important role, for example training on technical documentation for medical devices. These trainings promote an understanding of industry-specific standards and regulatory requirements and contribute to quality assurance.
- Are the documents in the dossier updated when new versions are released?
When a dossier is released, the version is ‘frozen’. Changes are only possible in a subsequent version. With the overview ‘Version statuses’ you get an overview of the document versions. To adopt new versions, create a new version of the dossier and decide which version of the documents to adopt.
For documents added to the dossier as attachments, the valid release version is always used.
- Can individual documents from the dossier be printed?
Printing of documents is possible, the function depends on the specifications from the document management system (controlled – not controlled).
- Is a partial export of the dossier possible?
Export as ZIP archive or PDF file is possible for the whole dossier or for selected chapters/documents.
- Is the data exported from the dossier protected?
For the ZIP file, the protection is preset with a password. Furthermore, you can preset the creation of PDF/A files to prevent printing and copying and enter a watermark for each export.
- Is the export logged?
The actions are recorded in the audit trail of the dossier, including the output as a ZIP or PDF file and the export of individual documents. The audit trail stores the date and time of an activity for the actions listed below. Moreover, the ID of the user triggering the activity is stored in the audit trail entry.
- Which file formats can be used in the dossier?
Documents can be inserted into the dossier regardless of the file format (PDF, MS Word, Excel, PowerPoint, PNG, JPG, TIFF, Visio).
Overview Medical Device Regulation (MDR)
The Medical Device Regulation (MDR) has been valid in the European Union (EU) since May 26, 2021. This article provides information on its content and the implementation provisions for the medical technology industry.
The “Regulation (EU) 2017/745 of the European Parliament and of the Council on medical devices” came into force on April 5, 2017. It replaces the previous “Directive 93/42/EEC on medical devices”. In concrete terms, this means that EU Regulation 2017/745 replaces the following previous directives in the field of medical technology:
- Medical Device Directive 93/42/EEC (MDD);
- Directive 90/385/EEC, active implantable medical devices – Active Implantable Medical Devices (AIMDD)
The EU bodies opted for a separate regulation in relation to Directive 98/79/EC on in vitro diagnostic medical devices (IVD). This directive has not been incorporated into the Medical Device Regulation. It will be replaced by the separately formulated EU regulation “In-vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR)”.
In contrast to the superseded directive, the EU Parliament’s new regulation has gained international significance. Its binding validity extends to all EU member states from May 26, 2021. It should be noted that no national legal acts had been adopted by the individual member states by October 2019. This also applies to special German features, requirements and regulations to be issued. Although there is a corresponding draft for a German implementation law, the Federal Ministry of Health expects transitional periods.
According to the current legal text, a transitional period of three years (26.05.17 – 25.05.21*) applies for implementation. Manufacturers have been obliged to submit an MDR certificate since the deadline expired at the latest. This is the only way they are allowed to launch a product on the market for the first time.
*In the wake of the COVID 19 pandemic, the effective date of the regulation was delayed to May 26, 2021
At its core, the new regulation aims to provide optimised, uniform rules for the market launch of medical technology products. The focus here is on product safety and product quality. For market participants and users, the new EU regulation consists of a mixture of familiar content from the previous Directive 93/42/EEC and far-reaching innovations. The main substantive and procedural aspects for you and your sector include the following contents of the Brussels Regulation:
Rules for the classification of medical devices
New terms and rules lead to changes in the allocation of existing products to product classes, some of which have changed. The field of stand-alone software products, for example, is affected by these changes.
Conformity evaluation procedure
In line with the classification regulations, the conformity assessment procedure now also differs depending on the class.
Technical Documentation/Document Management System
Technical documentation in accordance with the Medical Device Regulation proves to be much more detailed. The advantages for companies in terms of control and documentation become clear when you look at the new underlying, tabularly structured scheme in detail. The subdivision into individual categories in direct connection with the associated detailed requirements forms the basis for detailed technical documentation.
Clinical evaluations/clinical verifications
This action point is characterised by significantly higher requirements. This applies in particular to high-risk product groups.
Company responsibility/safety officer
A significant change for your company results from the requirement of the EU bodies for a responsible person who is responsible for compliance with the regulatory requirements in your organisation.
Quality management
The FDA Medical Device 21 CFR 820 (FDA = U.S. Food and Drug Administration) stipulates the requirements for management systems of medical device manufacturers. In this function, Device 21 CFR 820 is a counterpart to ISO 13485. The core requirement is that procedural instructions such as document control, procurement, development and production are documented and implemented analogously.
Market surveillance mechanisms
In particular, the mandatory use of the EUDAMED database ensures greater transparency. It is available to both the public and direct competitors as an information medium. The characteristics of each medical device are mapped using appropriate links. Supply chain traceability is also possible.
In the interest of timely compliance with the new regulation, you are the responsible person in the company. A well-timed and coherent plan for your company’s transition to the Medical Device Regulation is a prerequisite for your success. You should give high priority to the life cycles of your medical device portfolio. Existing MDD certifications will lose their validity on May 26, 2024 in accordance with EU regulations. When planning, please bear in mind that your products already certified according to MDD must also undergo the new conformity procedure to be created according to MDR. Grandfathering is not guaranteed.
During the changeover phase, you are particularly challenged in your role as manufacturer or supplier. The top priority is to designate and train the person responsible for compliance in your company (MDR Article 15). Updates to internal quality assurance procedures, documentation and the amended product classification are mandatory.
Against this background, the following standardised procedure is recommended according to the following 4 points:
- Carrying out the conformity assessment in relation to the product
- Issuing the declaration of conformity
- “CE” marking of the products
- Registration of the company and products in the EUDAMED database.
With the MDR, the EU authorities have set out to create a modern, standardised solution for the Quality Management and market release of medical devices. Great attention is paid to the quality and safety of the products. Perhaps you are one of the small and medium-sized companies in the medical technology industry that fear over-regulation due to the extensive regulations and additional inspection bodies?
In particular, the assessment of conformity by the “compliance manager” to be hired is likely to become a personnel problem in the start-up phase. In the event of hardship, a shortage of certified inspectors could delay the certification required for market release. Medical technology industry associations are concerned that a bottleneck in certification could have a negative impact on the market launch of innovative and marketable products.
Quick contact
You have a question about Technical Documentation?
Our sales team will help you promptly and gladly.