Definition of the term ("What is Chapter 4 of the EU GMP Guide?")
Chapter 4 of the EU GMP Guide focuses on documentation and records in pharmaceutical production. This section is crucial, as accurate documentation ensures that the manufacture of medicinal products meets the highest quality standards.
EU GMP Guide, Chapter 4: The significance of documentation
- The significance of documentation: Proper documentation is a cornerstone of any pharmaceutical manufacturing practice. It enables manufacturers to make the entire manufacturing process traceable and ensure that all steps are performed in accordance with valid regulations and standards. Chapter 4 of the EU GMP Guide is specifically dedicated to this important topic.
- The requirements of Chapter 4: Chapter 4 sets out detailed requirements for documentation in pharmaceutical production. This includes the creation, review and updating of documents such as manufacturing instructions, process instructions, records and protocols. Documentation must be accurate, complete and in an understandable form.
Compliance with Chapter 4 of the EU GMP Guideline
Compliance with the requirements of Chapter 4 is of critical significance to ensure conformity with EU GMP guidelines. Proper documentation is essential not only from a regulatory perspective, but also from a quality perspective. It helps to minimize errors, detect and correct deviations, and ensure the quality and integrity of manufactured medicinal products.
Your path to digitization - Discover our software
Our digitization solutions primarily address document-based processes in manufacturing, production and quality management. The basis of the dls | eQMS is a holistic ECM/DMS system. The ECM/DMS system can be connected to your existing ERP system (e.g. SAP) and thus map almost all document-based processes in the company.
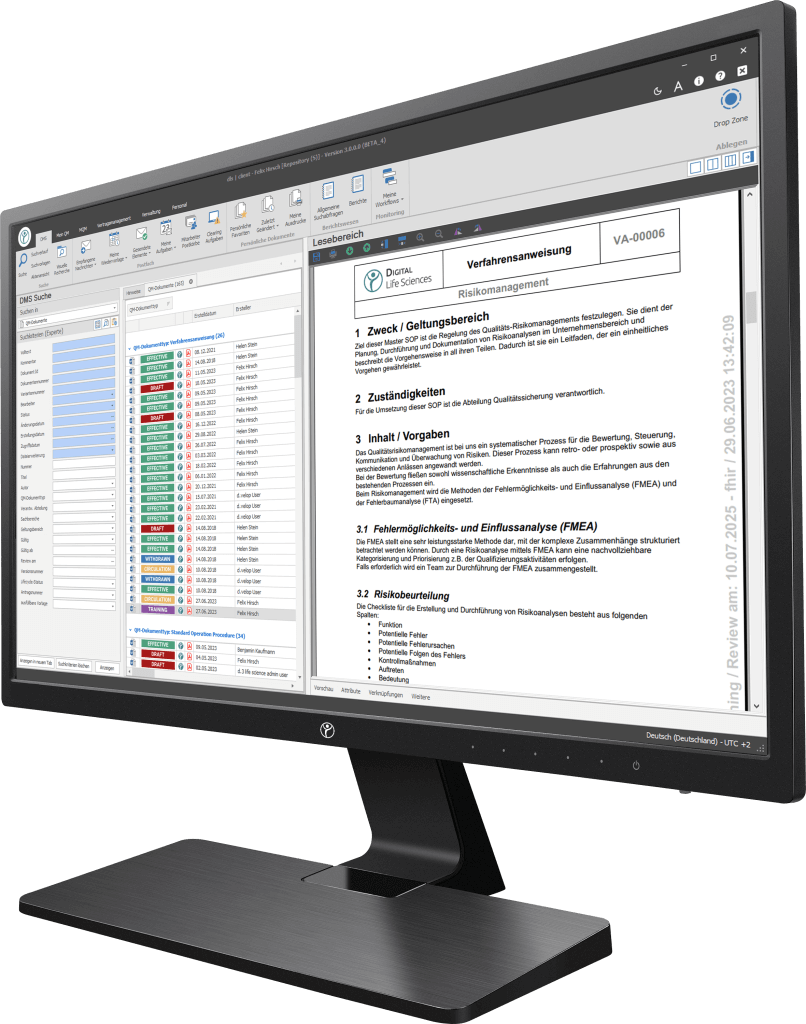
What are the documentation challenges?
Proper documentation management can be challenging. It requires careful planning, training of employees, and the use of appropriate software and technology. Organizations often face the challenge of managing large volumes of documents and ensuring that they are always up-to-date.
What are the important parts of the documentation?
- Document type: Batch documents are detailed documents that provide a chronological report of the manufacturing process of each batch. They contain information on raw materials, equipment used, procedures carried out and test results. These records are essential for traceability and quality control.
- Specifications: Specifications outline the quality parameters that a product must fulfill. They cover various aspects such as the identity, strength and clarity of the ingredients. Compliance with these specifications is crucial for ensuring product quality.
- Change control: Change control procedures are in place to manage modifications to processes, equipment or facilities. Chapter 4 of the EU GMP Guide emphasizes the need for a well-documented change control system for the evaluation and implementation of changes.
- Validation protocols: Pharmaceutical processes and equipment must be validated to ensure their reliability and consistency. This chapter emphasizes the significance of documenting validation protocols and the results achieved.
The role of records
Accurate and well-maintained records are not only a regulatory requirement, but a fundamental aspect of ensuring product quality and patient safety. Chapter 4 outlines the following key points regarding the records:
- Retention periods: The chapter sets out the minimum retention periods for different types of records. It is of crucial significance for manufacturers to adhere to these guidelines in order to facilitate inspections and audits.
- Data integrity: Data integrity is a crucial component of record keeping. Records must be accurate, complete and tamper-proof to prevent data manipulation or fraud.
Is the implementation of the EU GMP guidelines, chapter 4 mandatory?
Compliance with EU GMP Guideline, Chapter 4 is not optional, but mandatory for pharmaceutical manufacturers operating within the European Union. This way you can ensure effective implementation:
- Training: Make sure your personnel are well trained in documentation practices and understand the significance of accurate record keeping.
- Quality assurance: Establish a robust quality control system to review and approve all documents and records.
- Technology: Invest in modern document management systems and electronic records to optimize the documentation process.
- Auditing: Conduct regular internal audits to identify and correct deviations from the guidelines.
Conclusion
The EU GMP Guideline, Chapter 4 is an important chapter in the world of pharmaceutical manufacturing. It sets the standards for documentation, ensuring product quality, traceability and compliance with legal requirements. By following the guidelines and best practices outlined in this chapter, pharmaceutical companies can fulfill their obligation to bring safe and effective medicines to market.
Start your digital transformation with our powerful, modular software solutions
Frequently Asked Questions (FAQs)
What is the main objective of the EU GMP Guide, Chapter 4?
EU GMP Guidance, Chapter 4 aims to ensure that pharmaceutical manufacturing processes are well documented, standardized and in compliance with regulatory requirements.
How long should pharmaceutical records be kept in accordance with this chapter?
The chapter sets different retention periods for different types of records, but it is critical to follow them conscientiously. For example, batch documents should generally be kept for at least one year after the expiration date of the product.
Can electronic records completely replace paper-based records?
Although electronic records are recommended for efficiency, some paper-based records, such as batch documents, may still be necessary for certain processes. It is important to maintain the integrity and authenticity of both electronic and paper-based records.
What happens if a pharmaceutical company does not comply with EU GMP guidelines, chapter 4?
Non-compliance can have serious consequences, including regulatory measures, product recalls and damage to company reputation. Compliance is a top priority for pharmaceutical companies.
Are there resources to understand and implement this chapter?
Yes, regulatory authorities often provide guidance and training materials to assist pharmaceutical companies in understanding and implementing the EU GMP Guidance, Chapter 4.
How can a pharmaceutical company keep up to date with the latest revisions and updates to this chapter?
It is crucial to stay informed about updates to the EU GMP Guide, Chapter 4. Companies can regularly check the website of the relevant regulatory authority or visit industry associations for updates and training opportunities.
Which documents are required according to GMP in chapter 4?
In accordance with GMP, Chapter 4 lists the documents required by GMP that must be included in the documentation. This includes the site master file, regulations, logs and reports on actions in accordance with the regulations.
What should be defined in the manufacturer’s quality management system?
The various document types and the media used should be defined in the manufacturer’s quality management system.
What controls should be in place to ensure document accuracy?
Controls should be in place to ensure the accuracy, correctness, availability and legibility of the documents.
To which documents do the above aspects apply?
The aspects mentioned apply to both paper-based and electronic documentation systems as well as hybrid systems.
Why is electronic data often used for research purposes, even if paper printouts are available?
Electronic data is often used for research purposes, as paper printouts cannot usually be considered complete and electronic data contains additional information such as raw data and metadata.
What requirements apply to paper printouts with regard to electronic data?
Paper printouts must contain clear references that refer to the corresponding electronic data.
When is electronic batch release mandatory in accordance with Chapter 4?
An electronic batch release is mandatory in accordance with Chapter 4 if the data is primarily available in electronic form.
What are the requirements for complex systems, including software?
Complex systems, including software, must be validated and regularly checked, as prescribed in Chapter 4 of the EU GMP guidelines.
What needs to be considered when making changes or entries in documents?
Any changes or entries in a document must be signed and dated. The original information must be stored in such a way that it can be retrieved and the reason for the change must be logged.

